Archivo del Autor: Eduardo Chicano
SRM: La validación de resultados del siglo XXI
Hola de nuevo a todos, después de tanto tiempo, vuelvo a escribir un nuevo post.
Esta vez se lo dedico a una persona a la que admiro en divulgación científica. De hecho este año ha ganado el bitácoras en el apartado ciencia. Se trata de Jose, @scientiaJMLN, al cual, si no seguís tanto en su blog como en twitter ya os digo que estáis tardando. Jose, va por tí maestro!
Al grano, pues… Ya sabéis por el post anterior lo bien que me llevo con el Western Blot. Puede considerarse que el Western es para mi un archienemigo tipo Spiderman-Duende Verde, Sauron en el Señor de los Anillos, o cualquier super malo que se os ocurra.
Aun a pesar de todos los pesares, esta técnica se sigue utilizando a día de hoy como «estándar» para validación de resultados observados en los experimentos que realizamos los pocos locos que seguimos en España haciendo investigación. Ésta técnica como deberíais saber ya, es de los años 70, una época un poco lejana. Pero se sigue utilizando… y diréis todos ¿por qué? ¿No se ha avanzado nada desde esa fecha? Eso digo yo… y como buenos lectores que sois, os voy a explicar una técnica que puede servir (y de hecho bajo mi punto de vista ya debería ser estándar) para realizar el trabajo del Western más barato, más rápido, más específico y lo mejor de todo, de forma cuantitativa, no semicuantitativa como en el caso de mi archienemigo…
La técnica de la que os voy a hablar hoy es el SRM (Selected Reaction Monitoring). Se trata a grandes rasgos de poner un espectrómetro de masas a buscar y cuantificar una o varias proteínas de forma específica en una muestra donde tenemos varios miles de proteínas diferentes.
¡Ah! ¡Muy bien! Seguro que es súper fácil… diréis. Bueno, fácil no es. Ese es el concepto, lo que hay detrás es algo más complicado y es lo que quiero que a grandes rasgos entendáis para que al próximo que os presente al Western como herramienta de validación le déis en los morros con argumentos.
Empecemos. SRM, Selected Reaction Monitoring. Todos sabéis a estas alturas que las proteínas están compuestas por una secuencia de aminoácidos. Esas secuencias de aminoácidos son únicas para cada una de las proteínas que existen. La secuencia de una proteína puede ser «cortada» o digerida con determinadas enzimas (Leed esto). A partir de una digestión tríptica (esto es, con tripsina), la secuencia de aminoácidos de una proteína va a cortarse entre los aminoácidos Arginina y Lisina. De esta forma vamos a obtener una serie de fragmentos denominados péptidos los cuales, gracias a las características de esta enzima, van a ser digamos «estables» en diferentes muestras. Es decir, la tripsina siempre va a cortar en K-R (excepto si a K o R le sigue una P (prolina)), por lo que los péptidos obtenidos siempre van a ser los mismos aunque tengamos diferentes condiciones experimentales. Una vez que tenemos los péptidos de las proteínas, nos interesa conocer los niveles de expresión (o cantidad) de una o varias en concreto. ¿Cómo lo conseguimos? Pues seleccionando aquellos péptidos característicos de una proteína y cuya secuencia solo sea característica de esa proteína. Ese es el primer paso.
Resumiendo, tenemos una proteína cuya expresión queremos medir. Sabemos su secuencia, y la digerimos con tripsina para obtener péptidos de la misma. El péptido o péptidos con secuencia única y característica de esa proteína es lo que buscamos. Ahora quedaría otro paso más…
Normalmente, en un laboratorio de proteómica, se utiliza LC-MS para SRM (se puede hacer también con un Maldi, pero eso es otra cuestión que no viene al caso). Con LC-MS quiero decir que es una cromatografía líquida acoplada a espectrometría de masas.
El principal problema que vamos a encontrarnos si medimos con la premisa anterior una proteína es que cuando hagamos la cromatografía líquida, nuestro péptido de interés, el que caracteriza nuestra proteína, va a co-eluir junto a otros péptidos de secuencia diferente pero con la misma hidrofobicidad (se hace normalmente una fase reversa) y lo notaremos principalmente en su relación masa/carga, que es lo que vemos en el masas. Si esto ocurre, no podremos medir nuestra proteína de manera específica, ya que la señal que vamos a obtener va a estar «falseada» con los otros péptidos que no queremos medir. ¿Como solucionar esto y elegir solamente nuestro péptido y medir nuestra proteína? Pues volviendo a seleccionar otra característica de los péptidos: las transiciones o fragmentos originados con un espectrómetro de masas a partir de nuestro péptido de interés.
En los masas, como el triple cuadrupolo, una de sus «cámaras» puede utilizarse como celda de colisión de manera que el instrumento rompe los péptidos que entran en él en diferentes subfragmentos. No voy a entrar en como lo hace.
La cuestión es que esto lo vamos a utilizar de la siguiente forma: aplicamos un primer filtro en la primera fase de masas en la que seleccionamos la masa/carga (m/z) de nuestro péptido específico y con el cual pueden llegar a entrar algunos péptidos que nos den «ruido». Después pasamos esos péptidos con esa m/z a la celda de colisión donde los fragmentamos y en una segunda fase de masas (MS2) seleccionamos la m/z de aquellos fragmentos (transiciones) que sabemos que vienen de nuestro péptido de interés y ya medimos de forma muy específica lo que queremos: nuestra proteína.
Todo eso os lo resumo en la siguiente figura (Fuente Kinter et al 2013):

Como véis el flujo es sencillo: tenemos 3 péptidos entrando en el masas. El péptido A (el que nos interesa) tiene una m/z (masa/carga) de 370, por lo que ponemos el espectrómetro a seleccionar esa masa en la primera fase (MS1) de forma que solo nos dejará pasar a la siguiente fase los péptidos que tienen esa m/z. Con esto ya nos quitamos el péptido C, pero sin embargo, tenemos al péptido B que nos daría «ruido» en la medición de nuestra proteína.
Para ello pasamos todos los péptidos de m/z 370 a la celda de colisión y de todos los fragmentos generados solamente seleccionamos en una segunda fase (MS2) aquellos que provengan de nuestro péptido de interés (en azul) y más señal nos den y descartamos el resto. Así nos quitamos de enmedio el ruido provocado por el péptido B y sus fragmentos y fragmentos de nuestro péptido de interés que no nos sean útiles.
Todo este proceso se diseña en laboratorio bajo supervisión y no es tan sencillo como os lo explico aquí ya que hay que tener en cuenta multitud de factores, pero bueno, por lo menos así ya tenéis un arma más para matar a mi archienemigo.
¿Ventajas? Pues muchísimas: especificidad, cuantificación absoluta (no relativa como el Western), no se depende de una reacción de un anticuerpo, podemos medir hasta 100 proteínas (cuantificarlas, vamos) en un solo pase… etc, etc. Por no hablar de dinero, una cosa muy importante en tiempos de crisis. Creo que así a ojímetro, tras poner a punto el método puedes reducir los costes a una quinta parte de lo que cuesta un Western en igualdad de condiciones y además puedes hacer las mediciones más rápido, en cuestión de minutos.
Y lo mejor: una vez que diseñas el método de SRM te va a servir para toda la vida. Siempre es lo mismo. Y ya hay bases de datos en constante crecimiento donde los locos como yo vamos depositando esos métodos para que la comunidad científica los tenga a mano.
Espero que os haya entretenido aunque sea un rato ;-P
Digestiones e ionizaciones en el laboratorio
Antes de empezar con la serie de artículos interesantes creo que es necesario que sepáis algunos conceptos básicos que ocurren en el laboratorio antes de pasar a utilizar el espectrómetro de masas, como son la digestión y un par de tipos básicos de ionización.
Digestión
¿En que consiste exactamente una digestión cuando estamos en proteómica? Como sabéis las proteínas están hechas de una serie de piezas tal y como os puse en un post anterior. Cuando lo que queremos es identificarlas y saber la secuencia exacta en la que los aminoácidos están presentes en esa proteína utilizamos un espectrómetro de masas o secuenciación de Edman.
Actualmente, en proteómica tanto basada en geles como en líquido, se suele «cortar» esas proteínas en trozos más pequeños denominados péptidos para que el espectrómetro de masas que estemos utilizando sea capaz de darnos la secuencia de los mismos. De esta forma podemos saber rápidamente la secuencia o deducirla y podemos llegar a identificar la proteína o las proteínas en cuestión de minutos. Para ello comparamos las secuencias obtenidas con las bases de datos públicas disponibles en la red, siendo Uniprot una de las más utilizadas (bajo mi punto de vista, la mejor).
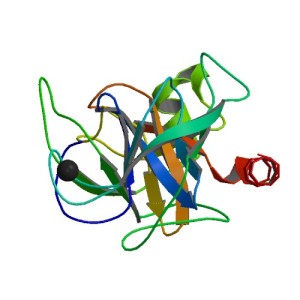
Tripsina (Fuente: RCSB)
Vale, todo esto está muy bien… ¿pero como se hace una digestión? En proteómica utilizamos una especie de «tijeras moleculares», unas enzimas (de la familia de las proteasas) las cuales están perfectamente caracterizadas tras años de estudio y de las cuales sabemos perfectamente su comportamiento.
La reina indiscutible para estos menesteres en proteómica es la tripsina. Esta enzima (que no encima, eso es arriba), rompe los enlaces peptídicos mediante una reacción de hidrólisis y se forman péptidos y aminoácidos a partir de una proteína completa. Normalmente la tripsina se produce de forma general en muchas células incluyendo entre sus localizaciones el cristalino, leucocitos, eritrocitos, piel, parótida, riñón e hipófisis, aunque es producida mayoritariamente en el páncreas y secretada en el digestivo a nivel del duodeno donde es esencial para la digestión que todos hacemos todos los días.
Esta proteína se puede purificar a partir de extractos de páncreas de cerdo y se compra comercialmente en casas especializadas en este tipo de productos (no, no voy a hacer publicidad gratuita, el que quiera saber alguna referencia que me pregunte, no hay problema 😉 ).
Bien, ya hemos hecho trizas nuestra proteína y la tenemos cortadita y picadita como si fuese un puerro, en juliana… Ahora toca prepararla para su análisis mediante espectrometría de masas.
Hago un pequeño paréntesis: normalmente la preparación de la muestra tanto antes de la digestión como después de la misma es un paso crítico para la obtención de unos resultados como Dios manda. Os lo recuerdo por si acaso no lo sabíais. Es más, una mala preparación pre-digestión o post-digestión os puede condenar al desastre en vuestro experimento si no lo habéis hecho con el máximo cuidado y esmero. Como responsable de un servicio de proteómica os digo que llega cada cosa a los servicios que os pondría los pelos de punta. Por favor, cuidad vuestras muestras. Intentad ser lo más cuidadosos posible y entregad las mismas a los servicios de proteómica en unas condiciones óptimas. De vuestro trabajo dependen en la mayoría de las ocasiones unos buenos resultados.

Columna de fase reversa C18. Observad la punta de la pipeta: esa cosita blanca que hay ahí es la resina donde se inmobilizan los péptidos
En fin, como os decía, tras la digestión, estas muestras suelen limpiarse tanto de restos de gel en su caso como de sales mediante la utilización de columnas de fase reversa,normalmente inmovilizadas en la punta de una micropipeta. Una columna no es más que una resina sintética que se coloca sobre esa punta de pipeta como si fuese una especie de filtro. La muestra (ya convertida en péptidos y aminoácidos) se pasa a través de esa columna, se retiene allí (interacciona con la resina) y se le pasan varias veces una serie de tampones para limpiar las sales y finalmente, se suelen eluir en un tampón compatible con nuestra fuente de ionización, dependiendo del espectrómetro de masas a utilizar, etc.
Fuentes de Ionización
En Proteómica se suelen utilizar principalmente fuentes de ionización bastante suaves. Yo os voy a hablar de la fuente de ionización tipo MALDI (Matrix-Assisted Laser Desorption/Ionization) que en cristiano viene a ser algo así como desorción/ionización láser asistida mediante matriz, y de la ionización tipo ESI (Electrospray Ionization).
Normalmente la fuente tipo MALDI se utiliza off-line, es decir, el espectrómetro que la utiliza no tiene ningún equipo asociado que permita el análisis continuado de muestra, como sí ocurre con la fuente tipo ESI, que es más común en espectrómetros de masas que tienen en cabecera on-line un nanoHPLC normalmente, lo que permite un análisis contínuo de muestra.

Placa MALDI donde se cocristalizan las muestras y la matriz
Mi especialidad es el MALDI, como os ireis dando cuenta con el tiempo. Este tipo de ionización se suele montar en un espectrómetro de masas que tiene como analizador un TOF o un TOF/TOF. No, no estoy tosiendo, jejejejeje. TOF significa Time Of Flight y lo que hace este tipo de analizador es separar las muestras que le entran mediante su relación masa/carga respecto al tiempo que tardan en recorrerlo. Pero bueno, ahora mismo esto no viene al caso, centrémonos en las fuentes de ionización.
La fuente MALDI consiste básicamente en lo siguiente: las muestras son coprecipitadas con una matriz, es decir, una sustancia usada para inmovilizar la muestra en un soporte metálico y que permitirá ionizar la muestra. El sólido resultante sobre el soporte se irradia con pulsos de láser con longitudes de onda en el infrarrojo o ultravioleta (normalmente láseres de N2 que emiten a 337 nm).
La matriz que se suele usar es casi siempre una molécula orgánica pequeña cuya absorbancia se encuentra dentro de la longitud de onda del láser que se esté usando. Las matrices difieren en la cantidad de energía que donan a las biomoléculas durante el proceso de desorción e ionización. El trabajo con biomoléculas casi siempre se realiza usando como matrices bien ácido α-ciano-4-hidroxicinnámico (CHCA) o bien ácido dihidrobenzoico (DHB).

Funcionamiento de una fuente de ionización MALDI (Fuente: http://www.magnet.fsu.edu)

Funcionamiento de una fuente de ionización ESI (Fuente: http://www.magnet.fsu.edu)
Con respecto a la fuente de ionización tipo ESI os diré que este método de ionización usa una aguja a cuyo través se bombean flujos muy reducidos de una disolución (nanolitros de una mezcla entre orgánico y agua, como metanol:agua, 50:50) con el analito disuelto. En su punta se aplica un voltaje muy alto por lo que el fluido se dispersa electrostáticamente en pequeñas gotas que se evaporan y dan su carga a las moléculas de analito que se ionizan a presión atmosférica. Para estabilizar el spray producido se suele usar un gas inerte. Una vez ionizado y estabilizado el spray, las moléculas de analito se transfieren al espectrómetro de masas con muy alta eficiencia.
Una vez ionizadas las moléculas de nuestra muestra, estas entran en los analizadores de los espectrómetros de masas que estemos utilizando, pero eso amigos míos, es otra historia que os contaré en otra ocasión.
Esta entrada participa en el XX Carnaval de Química organizado por @bioamara en su blog La ciencia de Amara.
Es la hora…

Jejejeje, que hay de nuevo viejo…
Maldito! Cuanto tiempo sin saber de tí y de tus actualizaciones de blog…
Cosas de la vida… querido visitante… te cuento…
Tras mi salida del Departamento de Bioquímica de la Universidad de Córdoba (cuanto me acuerdo de vosotros!), me he embarcado en una aventura en la que por ahora me encuentro cómodo, contento y porqué no decirlo, feliz. Entré en el IMIBIC hace ya unos meses y tras un inicio bastante estresante y un poco perdido (como cualquiera que empieza en un nuevo trabajo) ya me he habituado al nuevo ritmo de trabajo, que podría definirse como frenético, pero a la vez gratificante conforme vas viendo los frutos de tu trabajo.
Además, estoy aprendiendo un montón de cosas junto a mis nuevos compañeros, tanto los de las oficinas como los del edificio de Experimentación. ¡Qué manera de complicarse la vida tienen algunos! Eso sí… luego les salen unos experimentos que te quedas mudo…
También he iniciado una pequeña estancia en mi segunda casa en la UCO: la Unidad de Proteómica del SCAI. Allí estoy empezando a dar servicio al personal. Aprendiendo a gestionar una unidad, a realizar tratamiento de preparación de muestras, a manejar los distintos instrumentos que tienen allí disponibles… vamos, es como soltar a un crío en un chikipark de esos… me lo paso teta allí (pero trabajo duro, eh? que os creéis).
Bueno al grano, hoy el post va a ser cortito. Pienso darle un nuevo aire al blog e iré publicando de vez en cuando, a ver si me obligo porque si no @ScientiaJMLN, @bioamara, @lualnu10 y demás amiguetes de twitter y la blogosfera me van a matar (Os quiero chavales! Ya sabéis que os sigo en la sombra cuando puedo…). Mi objetivo en principio va a ser publicar comentarios acerca de artículos de interés que haya leído recientemente en Proteómica para que conozcáis que se cuece de primera mano en el frente de batalla y en perfecto cristiano. Nada de términos técnicos. Bueno, algunos sí, que ya están explicados en el blog y los voy a dar por sabidos.
Pues nada más, espero seguir por aquí todo lo que pueda molestando al personal. Para los que queráis más información de donde estoy trabajando y que es lo que podemos hacer os recomiendo una visita a la página del IMIBIC y a la de las UCAIB.
Un saludo y nos vemos pronto!

Una de Western
Excusas para explicar mi prolongada ausencia del blog tengo un montón, pero para que engañarnos, os he dejado de la mano de Dios y no hay perdón posible.
Entre trabajo (muchísimo), familia y las fechas que acaban de pasar, pues nada, el blog es el que pierde… Pero bueno, ya estamos aquí de nuevo con fuerzas renovadas y esperando que este año sea muchísimo mejor que el que ha pasado. A ver si es verdad.
Vamos al tema. El Western… mmmmm….. esa técnica que he aprendido a amar tanto como a odiar. A amarla porque cuando sale el experimento y lo tienes bien puesto a punto es una auténtica delicia, pero mientras la pones a punto o no, la odias a muerte cada vez que ves que el tiempo pasa y que los resultados no llegan. Os explico a mi manera (a nuestra manera) en lo que consiste esta técnica.

Imaginad por un momento que hacéis un experimento clásico de proteómica. Hacéis vuestro gel bidimensional, analizáis las imágenes, recortáis las manchas de interés de los geles e identificáis mediante espectrometría de masas la proteína que os está dando diferencia de expresión en vuestros análisis. Hasta ahí, todo correcto.
El siguiente paso sería la validación de los resultados y aquí es donde entra en juego nuestro amigo: El Western Blot.
El Western Blot (WB) es una técnica que nos sirve para detectar una proteína de interés de entre un conjunto complejo de las mismas. Para esa detección se utilizan anticuerpos específicos para esa proteína y después mediante el acoplamiento de distintos reactivos se puede llevar a cabo la visualización de esta proteína en esa mezcla.
Lo primero de todo para hacer un WB es separar la mezcla compleja de proteínas de partida con una electroforesis unidimensional. En este tipo de electroforesis separamos las proteínas únicamente por su tamaño. Las proteínas aparecerán a lo largo del gel de electroforesis formando bandas según su tamaño molecular.

Electroforesis unidimensional. (Fuente: Wikipedia)
A continuación se realiza una transferencia de estas proteínas separadas por tamaño a una membrana de nitrocelulosa o PVDF (Polifluoruro de vinilideno) donde quedarán inmovilizadas y donde se realizarán los tratamientos de detección. La membrana une proteínas de forma inespecífica lo cual podría representar un problema a la hora de realizar esa detección.
Montaje para llevar a cabo la transferencia a la membrana (Fuente: cultek.com)
Por ello, una vez que hemos realizado la transferencia se hace el bloqueo de la membrana. Imaginad que la membrana es como un papel en blanco donde hemos dibujado una serie de bandas, pero queremos rellenar todo este papel en blanco con color para que no nos quede ningún sitio libre donde se pueda dibujar. Eso es lo que hacemos con el bloqueo. El bloqueo se lleva a cabo utilizando leche desnatada, así, las proteínas de la leche nos rellenarán los huecos que han quedado sin proteína en la membrana para que nada más se una a ella.
Ya hemos realizado una electroforesis, una transferencia y hemos bloqueado la membrana. Ahora lo que tenemos que hacer es la hibridación con el anticuerpo primario.
El anticuerpo primario es un anticuerpo específico producido para reaccionar con nuestra proteína de interés. Este anticuerpo suele producirse en modelos animales (ratón, conejo, cabra, cultivos celulares…) y se purifica. Pero tranquilos, que no hace falta que purifiquemos nada. Actualmente hay muchas casas comerciales (no pienso hacer publicidad) que se dedican a «fabricar» anticuerpos contra todo tipo de proteínas que os podáis llegar a imaginar. Para producir estos anticuerpos lo que hacen es inocular a estos modelos animales la proteína completa o una parte de ella para que su cuerpo produzca anticuerpos contra esa proteína. Una vez que consiguen esto es cuando purifican ese anticuerpo y nos lo venden a precio de oro. Así a bote pronto, os puedo decir que el anticuerpo con el que estoy trabajando ahora cuesta unos 350 €/50ul. Sí, sí, 50 microlitros…
Sigamos… La hibridación con este anticuerpo primario nos permite, poner una «etiqueta» a nuestra proteína de interés y aquí es donde entra en juego la hibridación con el anticuerpo secundario.
Lo sé, lo sé, anticuerpo primario, anticuerpo secundario… ¡Vaya lío! Nada de eso. Ahora lo vemos.
Pensad por un momento que estamos utilizando un conjunto de proteínas de ratón, que hemos separado, transferido y bloqueado. Nuestra proteína de interés, vamos a poner por ejemplo, es la GAPDH (Gliceraldehído-3-fosfato deshidrogenasa). Entonces tendremos que buscar un anticuerpo primario anti-GAPDH de ratón. Ese anticuerpo puede proceder bien de modelos de ratón, de conejo, etc., pero para no complicar más la cosa vamos a poner que ese anticuerpo anti-GAPDH está «fabricado» en ratón. Hibridamos con nuestro anticuerpo primario.
Ahora que tenemos «la etiqueta» pegada a la GAPDH que tenemos en esas bandas de la membrana tenemos que buscarnos una forma de poder verla. Para eso se utiliza el anticuerpo secundario. Estos anticuerpos suelen estar modificados de forma que nos permitan utilizar métodos de detección enzimática (botina, peroxidasa de rábano, fosfatasa alcalina), radiactiva, fluorescente , etc.
Recordad que estamos trabajando con un anticuerpo primario de ratón, por lo que necesitaríamos un anticuerpo general anti-ratón que se una a nuestro anticuerpo primario de forma específica. En eso consiste la hibridación con el anticuerpo secundario, en pegarle multitud de anticuerpos modificados para la detección al anticuerpo primario y por ende para la detección de nuestra proteína.

Detección quimioluminiscente con peroxidasa de rábano (Fuente: Wikipedia)
Por último ya es cuestión de utilizar el método de revelado del WB que hayamos escogido con nuestro anticuerpo primario. En mi caso utilizo para la detección película fotográfica y luminol/peróxido (detección con peroxidasa de rábano) para poder ver esa proteína que estoy buscando.
Para una descripción más detallada de toda la técnica os recomiendo visitar la entrada del WB de la Wikipedia. Creo que lo resumen muy bien, pero seguro que no tan ameno como os lo he explicado yo.
Si tenéis alguna duda o pregunta que hacerme ya sabéis que estoy disponible a través de los comentarios en el blog o en mi twitter @educhicano.
Pues nada más, espero que os haya gustado esta entrada. Un saludo!

Hágase la luz
La luz… Una dualidad onda/corpúsculo que lleva con nosotros desde el comienzo de los tiempos… Y que también sirve, como no, para realizar análisis en Proteómica.
Así a bote pronto se me ha ocurrido explicaros 3 aplicaciones cotidianas en todo laboratorio de Proteomica que se precie que usan la luz como método de análisis o como parte de un análisis.

Luz láser. Fuente
La primera de estas técnicas es una de las mas citadas por todos los artículos proteómicos: la técnica de Bradford.
Esta técnica sirve para medir la cantidad de proteína presente en una muestra. Sí, lo se, lo se, hay mas métodos (por ejemplo, el método de Lowry) pero al fin y al cabo todos basan su medida final en el resultado que de un espectrofotómetro que va a medir la variación de Absorbancia de luz de una muestra.
En un ensayo de proteínas de Bradford se emplea un colorante hidrofóbico en una mezcla de agua y de ácido fosfórico. Esta mezcla tiene un color pardo-rojizo y suele comprarse en forma de preparado comercial.
La disolución del colorante al entrar en contacto con el interior de una proteína, vira su color de pardo-rojizo a azul. Este cambio de color se puede medir mediante el uso de un espectrofotómetro cuya fuente de emisión de luz se encuentre a 595nm (espectro visible).

Ensayo de Bradford. De izquierda a derecha con cantidades decrecientes de BSA. Fuente
Este método de medida de cantidad de proteína depende de la interacción entre este colorante hidrofóbico y las proteínas y es muy sensible a la presencia de contaminantes (detergentes, orgánicos…) por lo que hay que tener en cuenta como se ha preparado la extracción de proteínas que vamos a medir. Además tiene un rango lineal bajo, por lo que es necesario realizar disoluciones en muchas ocasiones para que la medida quede dentro del rango de medición.
Para superar estas limitaciones existen varias alternativas también basadas en las medidas mediante espectrofotometría y que suelen adquirirse bien preparadas o bien se fabrican de forma «casera» en el laboratorio siguiendo protocolos establecidos a tal efecto.
La principal ventaja del método de Bradford es que es muy fácil de llevar a cabo y muy rápido (a mi no me lleva más de 10-15 minutos) y cosa que no puedo decir de otros métodos alternativos que se me hacen largos y tediosos.
La segunda de las técnicas es la adquisición de imágenes a través de un escáner láser. Así es, podemos iluminar de forma un gel como los de la anterior entrada o cualquier otro teñido con un fluoróforo (Sypro, ProQ Diamond, etc), punto por punto y a la longitud de onda justa para que sólo ese fluoróforo se excite y nos emita luz y obtener una imagen con una calidad excepcional.

Escáner Typhoon de GEHealthcare
Por ultimo y no menos importante, la aplicación de la luz para iniciar el análisis de la secuencia de péptidos e identificación de proteínas. Para esto también utilizamos láser y en concreto lo hacemos para excitar una serie de moléculas de una sustancia que contiene nuestros péptidos. Es lo que se conoce como fuente de ionización Maldi.
La desorción de iones asistida por láser (MALDI: Matrix-assisted laser desorption ionization) es uno de los dos métodos de ionización “suaves” que actualmente se utilizan en espectrometría de masas. Fue desarrollado por Karas y Hillenkamp a finales de los 80.
Las muestras son coprecipitadas en un soporte metálico con una matriz, es decir, una sustancia usada para inmovilizar la muestra y que permitirá ionizar la muestra. Esta matriz suele ser ácido alfa-ciano-4-hidroxicinnámico, ácido sinapínico o ácido 2,5-dihidroxibenzoico.

Soporte metálico que se utiliza en una fuente de ionización MALDI. Fuente: Eduardo Chicano
Pues bien, el sólido resultante (recordad, mezcla de matriz y nuestros analitos) depositado sobre ese soporte se irradia con pulsos de láser con longitudes de onda en el infrarrojo o ultravioleta (normalmente láseres de N2 que emiten a 337 nm).
Para que os hagáis una idea, esto es lo que ocurre:

Funcionamiento de una fuente de ionización MALDI. Fuente
El láser provoca que la matriz se excite y literalmente «salte» del soporte, ionizándose de camino junto con nuestros péptidos, que es lo que nos interesa analizar , entrando en la segunda fase de un espectrómetro de masas, el analizador (tipo TOF, cuadrupolo, etc).
Pero la historia de los componentes y tipos de espectrómetros de masas os lo dejo para otro día…

Este Post participa en la VII edición del Carnaval de Biología, albergado por Manuel Sánchez en su blog Curiosidades de la Microbiología
PROTEÓMICA CLÍNICA: ASPECTOS BÁSICOS Y APLICACIONES

Nosotros también tenemos colores
Mis colegas que trabajan en Biología Celular tienen unas de las aplicaciones de proteínas más chulas y que más me han fascinado desde que entré en este inmenso mundo que es la Ciencia. Os confieso que la primera vez que pude ver una cosa así en directo en un microscopio confocal me quedé literalmente sin palabras y entendí la de vueltas que llegamos a darle a las cosas los científicos con tal de realizar experimentos útiles.

Gato con el gen de GFP insertado. Fuente: Xatakaciencia
Efectivamente, os hablo de las proteínas fluorescentes. Ellos utilizan generalmente tres tipos de proteínas (GFP, YFP y RFP) para poder ver «in vivo» los acontecimientos que están ocurriendo en la célula y que son de su interés. Recientemente habéis podido ver su utilidad para poder observar el desarrollo de una enfermedad similar al SIDA en gatos tal y como podéis ver arriba.
Pero espera… ¿que es GFP, YFP, RFP? Son los acrónimos de Green Fluorescent Protein (GFP), Yellow Fluorescent Protein (YFP) y Red Fluorescent Protein (RFP).
La GFP es una proteína que produce de forma natural la medusa Aequorea victoria. Esta proteína está constituida por 238 aminoácidos, los cuales forman once cadenas beta consecutivas que forman un cilindro en cuyo centro se encuentra una hélice alfa (espero que a estas alturas ya os hayáis leído la entrada de estructura del blog).

Estructura de la GFP. Fuente: Wikipedia
Esta proteína emite luz en la zona verde del espectro visible, por eso la vemos de color verde cuando iluminamos la célula/tejido/organismo con luz ultravioleta.
Esta proteína bioluminiscente se ha convertido en una herramienta clave en muchos de los experimentos de Biología Molecular y Celular desde hace varios años y nos ha permitido observar la expresión de genes y otras moléculas dentro de células u organismos completos. Las aplicaciones de la GFP son innumerables.
Como siempre, detrás de un gran descubrimiento suele haber grandes científicos que lo han llevado a cabo. De hecho, el descubrimiento, caracterización y aplicación de la GFP fueron el motivo por el que los estadounidenses Martin Chalfie y Roger Tsien y el japonés Osamu Shimomura compartieran el Premio Nobel de Química en 2008.
Osamu Shimomura fue la primera persona en aislar la GFP a partir de la Aequorea victoria e identificar qué parte era responsable de la fluorescencia. Martin Chalfie supo cómo utilizar la GFP para visualizar procesos en Biología Celular y por último, Roger Tsien, diseñó nuevas variantes de la GFP que brillan en distintos colores (YFP, RFP). Si queréis más información sobre aplicaciones que utilizan este tipo de proteínas no os olvidéis de visitar esta entrada del blog Jindetrés sal! del genial @DrLitos.

Utilización de diferentes proteínas fluorescentes en el marcaje de distintos compartimentos celulares. Fuente: Nikon
Es más, si lo que realmente queréis es la máxima información en profundidad acerca de la cantidad de variantes que existen y las propiedades de estas proteínas tan especiales os recomiendo este enlace (en inglés).
Pero no solo los chicos pijos de Biología Celular tienen colores para «jugar»… En clase, también los tenemos los proteómicos, aunque utilizando una aproximación un poco diferente.
En este caso os voy a explicar como conseguimos nosotros los colores con los que jugar. En definitiva se trata de añadirle a cada una de las muestras que estemos analizando el color que queremos.
Los proteómicos utilizamos para «teñir» las muestras (las proteínas) unos grupos químicos denominados cianinas que podemos unir a las lisinas que tienen las proteínas que estamos analizando.
La técnica con la que conseguimos los colores tiene como nombre DIGE (Differential In Gel Electrophoresis) y corrige los problemas de reproducibilidad, detección de manchas y posterior análisis de la 2-DE convencional. Se basa en las propiedades de 3 tipos de fluoróforos de la familia de las cianinas (CyDye: Cy3, Cy2, Cy5), que emiten luz a distinta longitud de onda.
Normalmente, los reactivos comerciales que se utilizan tienen un grupo reactivo (éster NHS) y están diseñados para formar un enlace covalente con el grupo amino (epsilon) de los aminoácidos de lisina de las proteínas vía unión amida. El fluoróforo añadido a la proteína de esta forma no nos va a varíar el pH de la proteína pero incrementa el tamaño de la misma sólo un poco y por eso se tiene en cuenta a la hora de analizar.
Bien, como os digo, cada muestra se marca con uno de los fluoróforos y, tras el marcaje, se mezclan antes del isoelectroenfoque. De esta forma se separan hasta 3 muestras diferentes (2 problema y 1 control generado al mezclar ambas, ó control interno) en un sólo gel, eliminándose la variación entre geles y asegurando que los efectos de sobre/sub-expresión visualizados en el gel se deben sólo a cambios biológicos. Así, cada mancha de proteína puede ser comparada fácilmente en distintas condiciones de manera semicuantitativa.

Esquema de un experimento DIGE clásico (Fuente: GEHealthcare)
Después de haber realizado la segunda dimensión de la electroforesis los geles con las 3 muestras se digitalizan mediante el uso de un escáner láser que excita a cada cianina en su longitud de onda correspondiente. Así obtenemos de cada gel 3 imágenes como estas:

Imagenes de un experimento DIGE obtenidas con un escáner láser. Fuente: Ludesi
Y una vez que se han obtenido nuestras muestras coloreadas podemos analizarlo mediante un software específico para ello y así poder comprobar que proteínas varían su expresión dependiendo de cada una de las muestras. Es tras el análisis cuando las escindiremos del gel y las analizaremos mediante espectrometría de masas.
¡No me puedo creer que quiten mi base de datos!
Bases de datos en Proteómica hay muchas. Y para lo que estemos buscando o analizando. Por ejemplo, si estamos buscando propiedades generales de proteínas que hemos identificado, podemos ir a Uniprot. Si queremos estudiar un poco más sobre los dominios de las proteínas en cuestión podemos visitar Pfam, Interpro… Por el contrario, si lo que queremos es predecir «in-silico» alguna modificación postraduccional en la secuencia de nuestra proteína deberíamos visitar el CBS danés.

Ya lo veis, hay bases de datos para «casi» todo. En general existen bajo mi punto de vista dos gigantes de las bases de datos: EBI (con sus correspondientes bases de datos y montón de aplicaciones) y NCBI (también con sus correspondientes chorrecientas aplicaciones).
Pero menos conocido es el formato de anotación que utilizan estas bases de datos para describir las propiedades de las diferentes proteínas que los científicos nos vamos encontrando a lo largo de años y años de experimentación. Hay anotaciones/bases de datos que son muy populares por este lado del mundo (Europa), como la que yo llamo anotación suiza, la UniprotKB (http://www.uniprot.org/uniprot/P00493), y otras igualmente populares en otros lugares (EEUU) como la anotación/base de datos tipo NCBI.(http://www.ncbi.nlm.nih.gov/protein/NP_038584.2).
Ambas bases de datos, y en general todas las bases de datos, utilizan diferentes «accession number», es decir, diferentes codificaciones para identificar la misma proteína dependiendo de la base de datos, el formato que se le de a las anotaciones y la información que contiene esa anotación o descripción de la proteína.
Para muestra, un botón. ¿Os habéis fijado en los dos enlaces anteriores que os he dejado? Seguro que no… 😉 Pues miradlos bien. Es la misma proteína con diferente accesión number (P00493 vs NP_038584.2) dependiendo de si nos encontramos ante una anotación tipo Uniprot o NCBI respectivamente.
Fijaos como dependiendo del accession number podemos tener más o menos información sobre la misma cosa. Para mi gusto es bastante más completo y con mejor estructura el de Uniprot, pero vamos, que si hay que utilizar el del NCBI tampoco le vamos a hacer ascos…
Recientemente he podido leer un artículo (Griss et al, Proteomics October 2011) que se plantea la desaparición del formato de anotación IPI (International Protein Index), una cosa que me afecta directamente a día de hoy ya que la base de datos que descargué en el Servicio de Proteómica de la Universidad de Córdoba (mi segundo laboratorio en la práctica) tiene todas las anotaciones en formato IPI y es con ella y con un programa de espectrometría de masas llamado ProteomeDiscoverer con los que me dedico a identificar proteínas del ratón de campo M. spretus y sus niveles de expresión mediante la técnica de iTRAQ que ya os explicaré en otro post.
Este formato de anotación tiene la estructura que podéis observar más abajo. Una serie de líneas en las que se nos muestra la identificación para la anotación IPI (ID), el accesion number que posee esa proteína en esta base de datos (AC), los cambios que ha sufrido esta entrada (DT), la descripción (DE), el organismo al que pertenece, en este caso el ratón M. musculus (OS), la clasifición taxonómica (OC), enlaces cruzados con otras bases de datos (DR), la secuencia al final, etc.
ID IPI00284806.8 IPI; PRT; 218 AA. AC IPI00284806; IPI00131306; IPI00281692; IPI00351673; IPI00381442; AC IPI00387495; IPI00626403; DT 15-MAY-2003 (IPI Mouse rel. 1.12, Created) DT 07-NOV-2007 (IPI Mouse rel. 3.36, Last sequence update) DE HYPOXANTHINE-GUANINE PHOSPHORIBOSYLTRANSFERASE. OS Mus musculus (Mouse). OC Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; OC Mammalia; Eutheria; Rodentia; Sciurognathi; Muridae; Murinae; Mus. OX NCBI_TaxID=10090; CC -!- GENE_LOCATION: Chr. X:50341314-50374836:1. DR UniProtKB/Swiss-Prot; P00493; HPRT_MOUSE; M. DR Vega; OTTMUSP00000018945; OTTMUSG00000017356; -. DR REFSEQ_VALIDATED; NP_038584; GI:96975138; -. DR UniProtKB/TrEMBL; B1B0W8; B1B0W8_MOUSE; -. DR UniProtKB/TrEMBL; Q6TDG6; Q6TDG6_MOUSE; -. DR UniProtKB/TrEMBL; Q99KF5; Q99KF5_MOUSE; -. DR ENSEMBL; ENSMUSP00000026723; ENSMUSG00000025630; -. DR UniParc; UPI0000003E3A; -; -. DR MGI; MGI:96217; Hprt; -. DR Entrez Gene; 15452; Hprt; -. DR UniGene; Mm.299381; -; -. DR CCDS; CCDS40972.1; -; -. DR trome; MTR002853; -; PRT. DR EPD; EP07058; MM_HPRT; Transferase. DR CleanEx; MM_HPRT1; -; -. DR InterPro; IPR005904; Hxn_phspho_trans. DR InterPro; IPR002375; Pr/py_Pribosyl_transf_CS. DR InterPro; IPR000836; PRibTrfase. DR Pfam; PF00156; Pribosyltran; 1. DR TIGRFAMs; TIGR01203; HGPRTase; 1. DR PROSITE; PS00103; PUR_PYR_PR_TRANSFER; 1. SQ SEQUENCE 218 AA; 24570 MW; 925CC0D4A6626E05 CRC64; MPTRSPSVVI SDDEPGYDLD LFCIPNHYAE DLEKVFIPHG LIMDRTERLA RDVMKEMGGH HIVALCVLKG GYKFFADLLD YIKALNRNSD RSIPMTVDFI RLKSYCNDQS TGDIKVIGGD DLSTLTGKNV LIVEDIIDTG KTMQTLLSLV KQYSPKMVKV ASLLVKRTSR SVGYRPDFVG FEIPDKFVVG YALDYNEYFR DLNHVCVISE TGKAKYKA
Lo que tenemos ante nosotros es un fichero de texto plano muy útil y con bastante información que puede utilizar rápidamente el programa en cuestión que estemos utilizando para realizar nuestras búsquedas, ya que este programa normalmente se va a fijar en la primera columna (ID, AC, DT, DE, OS, OX, etc) para realizar un índice de todas las proteínas de la base de datos que tenemos anotadas bajo este formato y después va a darnos los resultados en función de los parámetros de búsqueda que estemos fijando.
Como os digo, el artículo (publicado hoy mismo) analiza las consecuencias de no continuar con la base de datos IPI y para ello realiza distintos test en la esta base de datos y en la que va destinada a sustituirla (UniprotKB). Bueno, de hecho ya la ha sustituido ya que como veréis en el siguiente link del EBI, la base de datos IPI «se ha cerrado».
Estos científicos del EMBL (European Molecular Biology Laboratory) han realizado dos tipos de test de «mapeo» o búsqueda de las mismas proteínas sobre ambas bases de datos completas para comparar la coincidencia entre ambas bases de datos utilizando para ello la herramienta PICR (Protein Identifier Cross-Reference Service) del EBI.
En el primer test han utilizado PICR para comprobar la cantidad de referencias cruzadas que tenían las proteínas de IPI con respecto a otras bases de datos. Este test lo hicieron para comprobar cuantas entradas de la base IPI estaban representadas en la base UniprotKB a nivel de ID de proteína.
En el segundo test también utilizan PICR pero en esta ocasión utilizan como base de comparación que las secuencias de cada proteína sean idénticas entre ambas bases de datos para que no exista error de identificación entre ambas bases de datos.
Bien, pues lo que han averiguado me ha dejado perplejo a la par que un poco preocupado.
Con el primer test y la base IPI para humanos (la llamaremos IPIh) han descubierto que sólo el 70% de las proteínas se hallaban como ID única en UniprotKB, un 8% aproximadamente de la IPIh estaba relacionada con varias ID (mapeaba con varias ID) y un 21% no se encontraba (no había mapeo) en la base UniprotKB.
Con el segundo test (el de comparar homología de secuencias entre ambas bases de datos), el resultado fue que el 67% de las proteínas de la base IPIh se encontraba en la base UniprotKB con ID única, un 8% mapeaba con varias ID y un 25% directamente no mapeaba.
En el caso de los ratones, la base de datos IPI para ratón M. musculus (llámemosla IPIm), que es la base de datos que utilizo los resultados fueron idénticos tanto para el primer test como para el segundo, 78% mapeados con ID única, 12% con varias ID y 10% sin mapear.
Además de estos resultados con estos dos tests, este grupo de científicos realizó una digestión «in silico» de ambas bases de datos, de las cuales no os pongo los resultados porque son un poco más difíciles de ver, pero que me tranquilizaron aun menos…
Y preguntaréis, si bueno, ¿ y que? Pues la parte preocupante es que en la base IPIh tenemos entre un 21-25% de proteínas que literalmente perdemos como consecuencia de la sustitución de una base de datos por otra. Y en la IPIm tenemos un 10% de la base de datos que me va a desaparecer.
¿Consecuencias de esto? Pues que las identificaciones basadas en estos sets de la base de datos IPI que no puedan «mapearse» en Uniprot se perderán para análisis futuros (literalmente según el artículo), o lo que es lo mismo, se perderán en el olvido…
Esto nos afecta por ejemplo, si como es mi caso, estamos utilizando una base de datos «obsoleta» (bueno, defínanme obsoleta si la discontinuaron el mes pasado…) para (a nivel de espectrometría de masas) identificar proteínas que intervienen en nuestro experimento y queremos publicar los resultados. Los colegas que quieran repetir estos resultados tendrán que tener en cuenta este «handicap» ya que se podrán encontrar con que nosotros hemos identificado proteínas que supuestamente no existen al no estar incluídas dentro de la base de datos UniprotKB que es la actual y pueden acusarnos de dar datos erróneos.
Por eso, siempre, y digo siempre, os recomiendo tener una base de datos actualizada a la última versión cuando se trate de investigar cualquier tipo de análisis en proteómica y en general en cualquier campo en el que nos dispongamos a utilizar una base de datos.
Este Post participa en la VI edición del Carnaval de Biología, albergado por Copépodo en su blog Diario de un Copépodo.
Secuenciación de Edman
Volviendo la mirada hacia las entradas del blog, me he dado cuenta de que hay algunas cosas que asumo como que ya las conocéis y puede ser que no sea así. Por eso hoy vamos a explicar brevemente, o lo más brevemente posible como fue el origen de la secuenciación de las proteínas.
La secuenciación de proteínas fue (según mi humilde opinión) uno de los hitos históricos a tener en cuenta para las personas que trabajamos con las mismas ya que sin ella (y varias cosas más), probablemente no tendríamos Proteómica…

Pehr Victor Edman
Todo comienza con la labor de un joven científico llamado Pehr Victor Edman. En aquellas fechas se acababa de reconocer que las proteínas eran entidades con una estructura, una masa y una carga eléctrica definidas. Este hombre, como muchos de nosotros, se encontraba centrado en investigación básica en Estocolmo, en el Instituto Karolinska, haciendo su trabajo para su tesis doctoral cuando se le cruzó en medio una guerra. Era una época muy dura, y tras servir en el ejército sueco durante la Segunda Guerra Mundial continuó sus trabajos y en 1947 le concedieron un premio para ir al Rockefeller Institute of Medical Research en la prestigiosa Universidad de Princeton donde realizó sus trabajos de purificación y caracterización de la Angiotensina de sangre bovina.
En aquellos años colegas físico-químicos suecos habían descubierto que proteínas de diferentes tamaños moleculares poseían estructuras químicas específicas y más que probablemente individuales.Edman estaba muy interesado en la determinación detallada de la estructura de las proteínas y con su método consiguió dar lugar a una explicación estructural de sus diversas actividades biológicas.
Para su doctorado, Edman aisló de la sangre bovina una proteína pequeña que regula la presión arterial (Angiotensina), pero pronto se dio cuenta de que las proteínas están constituidas por pequeños «bloques o ladrillos»: los aminoácidos. Para dar una explicación estructural a las diversas actividades biológicas de las proteínas debía encontrar un método que le permitiera una determinación de la estructura de las mismas.
A Edman se le ocurrió la fantástica idea de una serie de reacciones basadas en el acoplamiento del fenilisotiocianato (PITC) con una proteína purificada y el uso de hidrólisis ácida. Esta serie de reacciones le permitieron analizar las proteínas de forma secuencial,aminoácido por aminoácido, y así preservar la información lineal estructurales necesarias para interpretar su actividad biológica.
El procedimiento de la degradación de Edman o secuenciación de Edman marca y elimina sólo el residuo N-terminal de un polipéptido, dejando el resto de enlaces peptídicos intactos. Para ello, se hace reaccionar el péptido con PITC eliminándose el residuo N-terminal en forma de derivado de fenilhidantoína (PTH). Después de la eliminación e identificación del residuo N-terminal mediante HPLC, el nuevo residuo N-terminal puede ser marcado, eliminado e identificado por repetición de la misma serie de reacciones. Este procedimiento se repite hasta que se ha determinado toda la secuencia.

Esquema de la Secuenciación de Edman
Edman ideó en la Universidad de Lund las condiciones de reacción adecuadas para todos los aminoácidos y la gran mayoría de las clases de proteínas además de minimizar el número de reacciones secundarias no deseadas.
Debido a que se tienen que realizar muchos pasos de reacción y muchas determinaciones, normalmente hoy día éste método se lleva a cabo mediante analizadores automáticos o secuenciadores que mezclan los reactivos en las proporciones adecuadas, separa los productos, los identifica y registra los resultados. Visionario en su campo, Edman diseñó en 1961 un instrumento automático llamado «secuenciador de proteína para determinar la estructura de las proteínas mediante el análisis de la secuencia de aminoácidos», el primer secuenciador automatizado de proteínas.
Edman continuó hasta su muerte trabajando para mejorar este método y así poder determinar cadenas más largas pero con menor material de partida.
Este Post participa en la VI edición del Carnaval de Biología, albergado por Copépodo en su blog Diario de un Copépodo.

Mapas de proteínas
Comenzamos con este artículo ya a entrar en materia y dejamos atrás los artículos básicos sobre aminoácidos, enlaces peptídicos y estructura de proteínas.
En el análisis global de los componentes de un proteoma nos podemos encontrar a grandes rasgos con dos estrategias:
a) Convencional: separar todas las proteínas de la muestra (por electroforesis en dos dimensiones en general) e identificar aquellas de interés por espectrometría de masas.
b) A gran escala (“shot-gun”): la muestra es tratada con una enzima proteolítica y los péptidos resultantes se separan por HPLC acoplada a un sistema de espectrometría de masas. Esto permite la identificación de todas las proteínas presentes en la mezcla.
En este post vamos a ver la primera de ellas, la estrategia convencional, la cual se basa en el empleo de la electroforesis bidimensional.
La electroforesis bidimensional es una técnica de alta resolución para separar mezclas de proteínas muy complejas. La base de su gran poder de resolución es la bidimensionalidad, es decir, que las proteínas son separadas ortogonalmente al aplicar dos métodos físicos distintos. Una primera dimensión separa las proteínas en un gel de acrilamida con gradiente de pH por su punto isoeléctrico (isoelectroenfoque, del inglés isoelectric focusing IEF). En la segunda dimensión las proteínas son separadas por su masa molecular por electroforesis en poliacrilamida en presencia de SDS (SDS-PAGE). Tras teñir el gel, las proteínas aparecen como manchas circulares. (www.bioinformatica.uab.es).
El punto isoeléctrico (pI) de una proteína es el pH al que su carga neta es cero, por lo que no se desplaza en un campo eléctrico. Al introducir una mezcla de proteínas en un gel con un gradiente de pH, sus extremos N y C terminal (recordadlo en este post) y sus residuos ionizables captan o liberan protones de acuerdo con el pH, por lo que su carga eléctrica global dependerá del pH del entorno. Al aplicar un campo eléctrico, todas las moléculas con carga neta positiva serán atraídas hacia el cátodo, y aquellas con carga neta negativa hacia el ánodo. A medida que las moléculas de proteína se acercan a su pI van perdiendo carga eléctrica, hasta llegar al valor de pH en que su carga neta sea cero y dejen de moverse. Se dice entonces que las proteínas han enfocado, pues todas las moléculas de una proteína inicialmente dispersas por todo el gradiente de pH se han concentrado en un solo punto, que corresponde a su pI; de aquí el nombre de IEF. El enfoque se realiza a voltajes muy altos, de hasta 10000V, y en condiciones desnaturalizantes (ej. 8M urea) para obtener la máxima resolución.
En sus inicios, a finales de los 70, estos gradientes de pH se generaban con una mezcla compleja de pequeños polímeros con gran capacidad tampón cerca de su pI, o carrier ampholytes (CA), aunque tenían problemas como distorsión del gradiente de pH, variabilidad entre geles y gran complejidad química. Hoy, los anfolitos han sido sustituidos por las tiras IPG (gradientes de pH inmovilizados en un soporte plástico) generados por la copolimerización, junto a acrilamida y bisacrilamida, de un gradiente de monómeros de acrilamida derivatizados con grupos ácidos o básicos cuya unión covalente a la matriz impide la distorsión del gradiente. Las tiras IPG tienen muchas ventajas: i) tolerancia a presencia de sales en la muestra, ii) mayor capacidad de carga, iii) numerosos gradientes de pH (incluso muy estrechos, de <1 unidad), iv) gran disponibilidad comercial (GEHealthcare, Bio-Rad Laboratories).
La segunda dimensión consiste en una separación SDS-PAGE convencional. La tira IPG se deposita sobre el gel SDS-PAGE y se lleva a cabo la electroforesis.

Esquema de una electroforesis bidimensional (2-DE)
Tras realizar la segunda dimensión hay que teñir los geles. Hay muchos métodos de tinción de proteínas en geles de poliacrilamida pero sólo algunos se usan en 2-DE. El más usado durante muchos años ha sido la tinción con plata, por su alta sensibilidad, pues revela menos de 1 ng de proteína. Sin embargo, la plata no es una tinción a punto final y su rango lineal es muy estrecho. Por ello, ha sido sustituido por métodos de tinción con colorantes fluorescentes como el Sypro Ruby que, aunque mucho más caros, tienen una sensibilidad comparable a la de la plata y un rango dinámico lineal de hasta tres órdenes de magnitud, óptimos para estudios cuantitativos.
Como desarrollo de la 2-DE ha surgido una nueva tecnología que saca todo el provecho de la 2-DE y la tinción con fluoróforos: la técnica DIGE (Difference Gel Electrophoresis). Esta técnica corrige los problemas de reproducibilidad, detección de manchas y posterior análisis de la 2-DE convencional. Se basa en las propiedades de 3 tipos de fluoróforos de la familia de las cianinas (CyDye: Cy3, Cy2, Cy5), que emiten luz a distinta longitud de onda. Cada muestra se marca con uno de ellos y, tras el marcaje, se mezclan antes del isoelectroenfoque; se separan hasta 3 muestras diferentes (2 problema y 1 control generado al mezclar ambas, ó control interno) en un sólo gel, eliminándose la variación entre geles y asegurando que los efectos de sobre/sub-expresión visualizados en el gel se deben sólo a cambios biológicos. Así, cada mancha de proteína puede ser comparada fácilmente en distintas condiciones de manera semicuantitativa.

Esquema de un experimento con técnica DIGE
Al final de todo el proceso de la electroforesis bidimensional vamos a obtener unos mapas de las proteínas que se estaban expresando en un determinado momento en el organismo/tejido/cultivo tan chulos como este que os muestro a continuación y que obtuvo el que os escribe durante el desarrollo de su Tesis Doctoral:

Mapa proteico real
Y aquí es donde comienza el trabajo de espectrometría de masas y bioinformática, pero eso ya es otra historia que os contaré otro día…

{kind=link}